Viral Mutation Analysis¶
known_mutations Heatmap
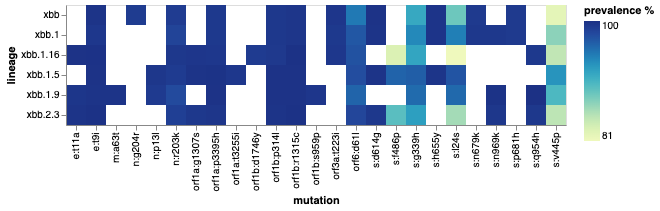
A basic but important question: how do we define a lineage? What mutations consistently appear in most sequences within a lineage? We could start by plotting some of the characteristic mutations of XBB occurring in 80% of sequences. Even better, we can do this for mulitple related lineages and compare them using a heatmap:
# Perform authentication if you haven't already
from outbreak_data import authenticate_user
authenticate_user.authenticate_new_user()
# Import outbreak_data package
from outbreak_data import outbreak_data as od
# Collecting 15 characteristic mutations and their prevalences for each variant
lin0 = od.known_mutations("xbb", freq = 0.80)
lin0["lineage"] = "xbb"
lin0 = lin0.iloc[:15]
lin1 = od.known_mutations("xbb.1", freq = 0.80)
lin1["lineage"] = "xbb.1"
lin1 = lin1.iloc[:15]
lin2 = od.known_mutations("xbb.1.5", freq = 0.80)
lin2["lineage"] = "xbb.1.5"
lin2 = lin2.iloc[:15]
lin3 = od.known_mutations("xbb.1.16", freq = 0.80)
lin3["lineage"] = "xbb.1.16"
lin3 = lin3.iloc[:15]
lin4 = od.known_mutations("xbb.1.9", freq = 0.80)
lin4["lineage"] = "xbb.1.9"
lin4 = lin4.iloc[:15]
lin5 = od.known_mutations("xbb.2.3", freq = 0.80)
lin5["lineage"] = "xbb.2.3"
lin5 = lin5.iloc[:15]
# Link each of the findings together for plotting
df = pd.concat([lin0, lin1, lin2, lin3, lin4, lin5])
df["prevalence"] = df["prevalence"].apply(lambda x: x*100)
df= df.rename(columns={'prevalence': 'prevalence %'})
# Plot mutation heatmap
import altair as alt
alt.Chart(df).mark_rect().encode(
x = "mutation:N",
y = "lineage:N",
color = 'prevalence %:Q')
mutation mutation_count lineage_count lineage gene ref_aa alt_aa \
0 orf6:d61l 4198 4423 xbb ORF6 D L
1 s:l24s 3919 4423 xbb S L S
2 n:r203k 4378 4423 xbb N R K
3 s:g339h 4054 4423 xbb S G H
4 s:v445p 3643 4423 xbb S V P
... ... ... ... ... ... ... ...
10 orf1a:p3395h 3263 3279 xbb.2.3 ORF1a P H
11 s:n969k 3261 3279 xbb.2.3 S N K
12 orf1a:g1307s 3257 3279 xbb.2.3 ORF1a G S
13 s:d614g 3257 3279 xbb.2.3 S D G
14 s:q954h 3257 3279 xbb.2.3 S Q H
codon_num codon_end type prevalence % change_length_nt
0 61 None substitution 94.912955 None
1 24 None substitution 88.605019 None
2 203 None substitution 98.982591 None
3 339 None substitution 91.657246 None
4 445 None substitution 82.364911 None
... ... ... ... ... ...
10 3395 None substitution 99.512046 None
11 969 None substitution 99.451052 None
12 1307 None substitution 99.329064 None
13 614 None substitution 99.329064 None
14 954 None substitution 99.329064 None
[90 rows x 12 columns]
Comparing Lineages
To better understand the evolution of SARS-CoV-2, we may also want to know what mutations are shared between different viral lineages and how many shared mutations there are.
For this example we'll look at three different lineages: B.1.1.7, P.1, and B.1.1.529. First, let's get an idea of which mutations are shared between all three lineages:
lin1 = od.known_mutations("b.1.1.7")
lin1["lineage"] = "b.1.1.7"
lin2 = od.known_mutations("P.1")
lin2["lineage"] = "P.1"
lin3 = od.known_mutations("b.1.1.529")
lin3["lineage"] = "b.1.1.529"
#Finding intersections
#Created an optional but convenient intersecting function
def intersect(lst1, lst2):
return set(lst1).intersection(lst2)
a = set(lin1['mutation'])
b = set(lin2['mutation'])
c = set(lin3['mutation'])
m12 = intersect(a, b)
m13 = intersect(a, c)
m23 = intersect(b, c)
m123 = intersect(m12, c)
print('Set 1.2:', m12)
print('Set 1.3:', m13)
print('Set 2.3:' , m23)
print('Set 1.2.3:', m123)
Set 1.2: {'orf1b:p314l', 'orf8:s84l', 'orf1a:del3675/3677', 'n:r203k', 's:d614g', 's:n501y', 'n:g204r'}
Set 1.3: {'s:p681h', 'orf1b:p314l', 'orf8:s84l', 'n:r203k', 's:d614g', 'n:g204r'}
Set 2.3: {'orf1b:p314l', 's:h655y', 'orf8:s84l', 'n:r203k', 's:d614g', 'n:g204r'}
Set 1.2.3: {'orf1b:p314l', 'orf8:s84l', 'n:r203k', 's:d614g', 'n:g204r'}
Finally, we can make a visual based on our findings and create a Venn diagram of our results:
# Import libraries for venn diagram visual
from matplotlib_venn import venn3, venn3_circles
from matplotlib import pyplot as plt
# Using matplotlib_venn to make venn diagram with custom visuals
## The venn diagram package used here automatically determines the number
## of intersecting values and plots accordingly
vd3 = venn3([a, b, c], set_labels = ('Alpha/B.1.1.7', 'Gamma/P.1', 'Omicron/B.1.1.529'), set_colors=('#c4e6ff', '#F4ACB7','#9D8189'),
alpha = 0.8)
c = venn3_circles([a, b, c,], linestyle = '-.', linewidth=2, color='grey')
for text in vd3.set_labels:
text.set_fontsize(16);
for text in vd3.subset_labels:
text.set_fontsize(16)
plt.title('Number of Overlapping Mutations in 3 Variants',fontname ='Helvetica',fontweight ='bold',fontsize = 18,
pad = 30,backgroundcolor ='#cbe7e3',color ='black', style ='normal');
c[0].set_lw(7.0)
c[0].set_ls(':')
plt.show()
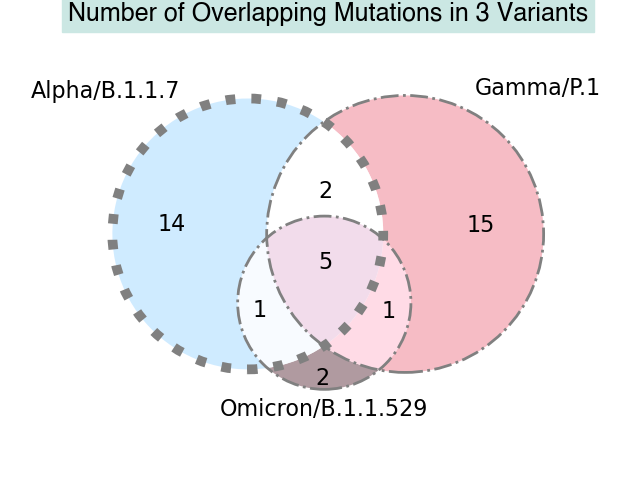
We can see that the graph matches the number of intersecting mutations that we previously saw, given that the venn diagram also takes into account unique values for each set.
Note
Please see the matplotlib venn diagram documentation and this article for more info on how to create these graphs.